
Os ensaios de PCR em Tempo Real são muito mais sensíveis, específicos e rápidos, principalmente quando comparados aos testes convencionais, levando de 2 a 3 horas para emitir o resultado.
Para diagnósticos, são amplamente utilizados na infectologia clínica para a detecção de patógenos, identificando infecções virais e bacterianas, em que a cultura dos agentes causadores pode ser muito difícil ou até mesmo impossível. Este método não depende do isolamento ou crescimento do patógeno ou da detecção de uma resposta imune contra o agente.
Em vez disso, o que é detectado nos ensaios são as sequências de ácidos nucleicos dos patógenos. Alguns exemplos de sua aplicação consistem no diagnóstico de dengue, infecções respiratórias, ISTs, anteriormente chamada de DSTs, e meningites.
É utilizada também no diagnóstico de doenças genéticas, identificando mutações e pré-disposição genética para determinadas doenças, como é o caso da trombose.
Essa técnica molecular também é utilizada na oncologia para identificação do cromossomo Philadeplhia, uma alteração citogenética que está associada a algumas formas de leucemia.
Metodologias:
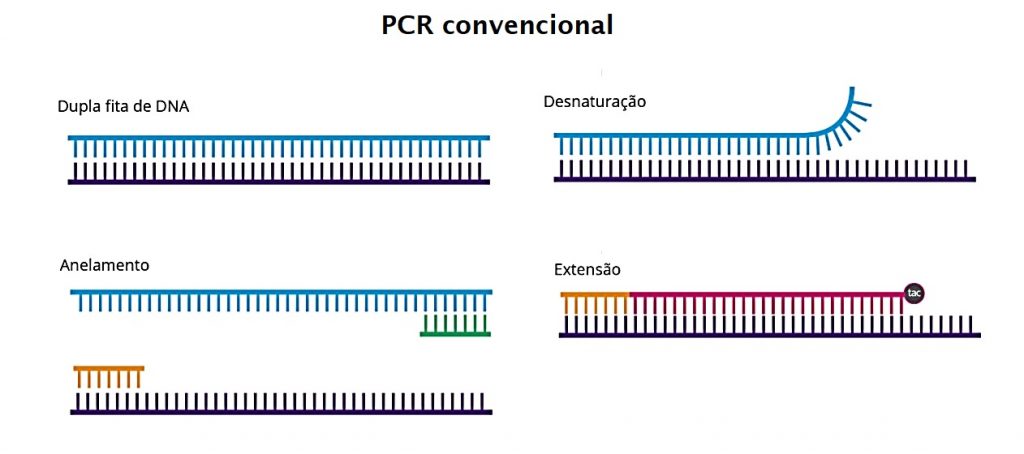
PCR CONVENCIONAL
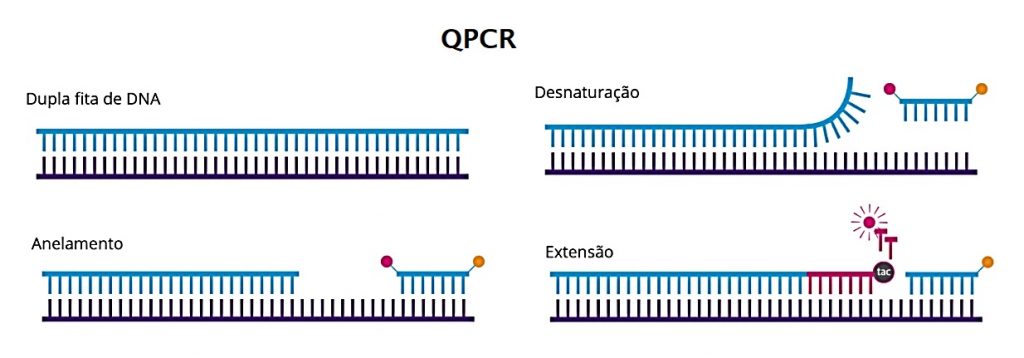
QPCR – PCR EM TEMPO REAL
PCR CONVENCIONAL
Existem 3 etapas que compreendem a reação de PCR:
- Desnaturação – O DNA gnômico contendo a sequência a ser amplificada é desnaturado. Ou seja, ocorre a separação da dupla fita de DNA.
- Anelamento ou Hibridização – os iniciadores ou primers se ligam a fita de DNA que se pretende amplificar. Um deles é complementar à sequência em uma fita da dupla-hélice de DNA e o outro é complementar à sequência na outra fita. São eles que irão identificar/marcar qual trecho de interesse do DNA deverá ser copiado.
- Extensão ou polimerização – com o ponto de partida já identificado, a Taq polimerase liga-se a fita sinalizada pelo primer, complementando-a. Inicia-se então a extensão do novo fragmento de DNA, formando novamente uma fita dupla de DNA.
Esse ciclo é realizado inúmeras vezes até atingir milhões de cópias. Na PCR convencional, a detecção do produto de amplificação normalmente é feita em eletroforese em gel de agarose. Após a coloração, ocorre a visualização do DNA pesquisado.
QPCR – PCR EM TEMPO REAL
Na qPCR, o resultado é visualizado imediatamente, dispensando a eletroforese. Isto é possível pela adição de sondas fluorescentes às reações de PCR. A amplificação do DNA-alvo é monitorada durante o processo de qPCR. A medida que o DNA é amplificado, o nível de fluorescência cresce proporcionalmente.
O equipamento é capaz de detectar a fluorescência eventualmente produzida pela amostra e assim a técnica permite acompanhar a reação e a apresentação dos resultados em tempo real.
Além disso, por meio do monitoramento da taxa de aumento da fluorescência na reação de PCR, é possível determinar com precisão a quantidade de DNA-alvo presente na amostra original. A qPCR pode ser utilizada para se avaliar a presença de um patógeno em uma amostra, podendo ser um teste qualitativo ou quantitativo.
INTERFERENTES NA TÉCNICA DE QPCR
A elevada sensibilidade dessa técnica possibilita excelentes resultados, porém também está sujeita a presença de inibidores e contaminação que podem afetar a sua eficiência. Os plásticos para qPCR tem um grande impacto sobre os ensaios em tempo real e precisam receber um tratamento especial para não interferir na reação, principalmente na leitura do sinal fluorescente. Ocasionam assim um resultado errado, com baixa concentração ou mesmo inviabilizando o processo.
Visualizar os resultados da PCR: através da eletroforese em gel
Principais acontecimentos e a evolução dessa técnica:
- 1983 – Criação da técnica por Kary Mullis.
- 1985 – Apresentação da pesquisa à comunidade científica e publicação na revista “Science”.
- 1986 – Taq polimerase – a descoberta da bactéria Termus aquaticus e extração de sua DNA polimerase possibilitou o aperfeiçoamento da técnica, que não necessitava de adição de mais DNA polimerase a cada ciclo, pois a mesma suporta temperaturas acima de 100°C.
- 1987 – Com a patente da PCR por parte da Perkin, desenvolve-se a automação para controle do aumento e redução da temperatura, neste momento surge o Temociclador.
- 1992 – Primeiro teste diagnóstico desenvolvido para HIV, descartando o problema da janela imunológica estabelecida pelo vírus.
- 1993 – Karry Mullis recebe o Prêmio Nobel de Química.
- 2003 – Desenvolvimento da PCR em Tempo Real ou qPCR– técnica de PCR na qual utiliza-se além dos primers (iniciadores de amplificação), as sondas marcadas, possibilitando a quantificação do alvo em estudo.
Referências bibliográficas disponíveis
Arnaldo, ZAHA, FERREIRA, Henrique Bunselmeyer, and PASSAGLIA, Luciane M. P. – organizadores. Biologia Molecular Básica, 4ª edição. ArtMed, 2012.
MADIGAN, Michael T., MARTINKO, John M., BENDER, Kelly S., BUCKLEY, Daniel H., STAHL, David A. Microbiologia de Brock, 14ª edição . ArtMed, 2016.
PINTO, Terezinha de Andreoli, KANEKO, Telma Mary, PINTO, Antonio F. Controle Biológico de Qualidade de Produtos Farmacêuticos, Correlatos e Cosméticos, 4ª edição . Manole, 2015.
MARTINS, Mílton Arruda, CARRILHO, Flair José, ALVES, Venâncio Ferreira, CASTILHO, Euclides. Clínica Médica, Volume 3: Doenças Hematológicas, Oncologia, Doenças Renais, 2ª edição . Manole, 2016.
Estamos a disposição. Nossa equipe técnica tem muito prazer em orientar e esclarecer a todos.
Célia Wada

Um comentário
Todas as aplicações citadas são para a PCR convencional e em tempo real ou só para a segunda?
Ana
Boa tarde
Me desculpe mas não entendi sua pergunta
Pode mandar via e-mail que te respondo – celia.wada@gmail.com
att